2026
Programming Biomolecular Interactions with All-Atom Generative Model
Xiangzhe Kong*, Junwei Chen*, Ziting Zhang*, Gaodeng Li*, Qingyuan Zhu, Lei Wei, Mingyu Li, Yan Shi, Weiyang Dai, Zishen Zhang, Wenjuan Tan, Rui Jiao, Xiaolun Wang, Jiqing Zheng, Ziyang Yu, Qilong Wu, Zhiye Guo, Li Zhang, Wentao Li, Qiaojing Huang, Xiaowo Wang, Wenbing Huang, Yuli She, Jian Zhang, Yang Liu, Kai Liu, Jianzhu Ma (* equal contribution)
Under review. 2026
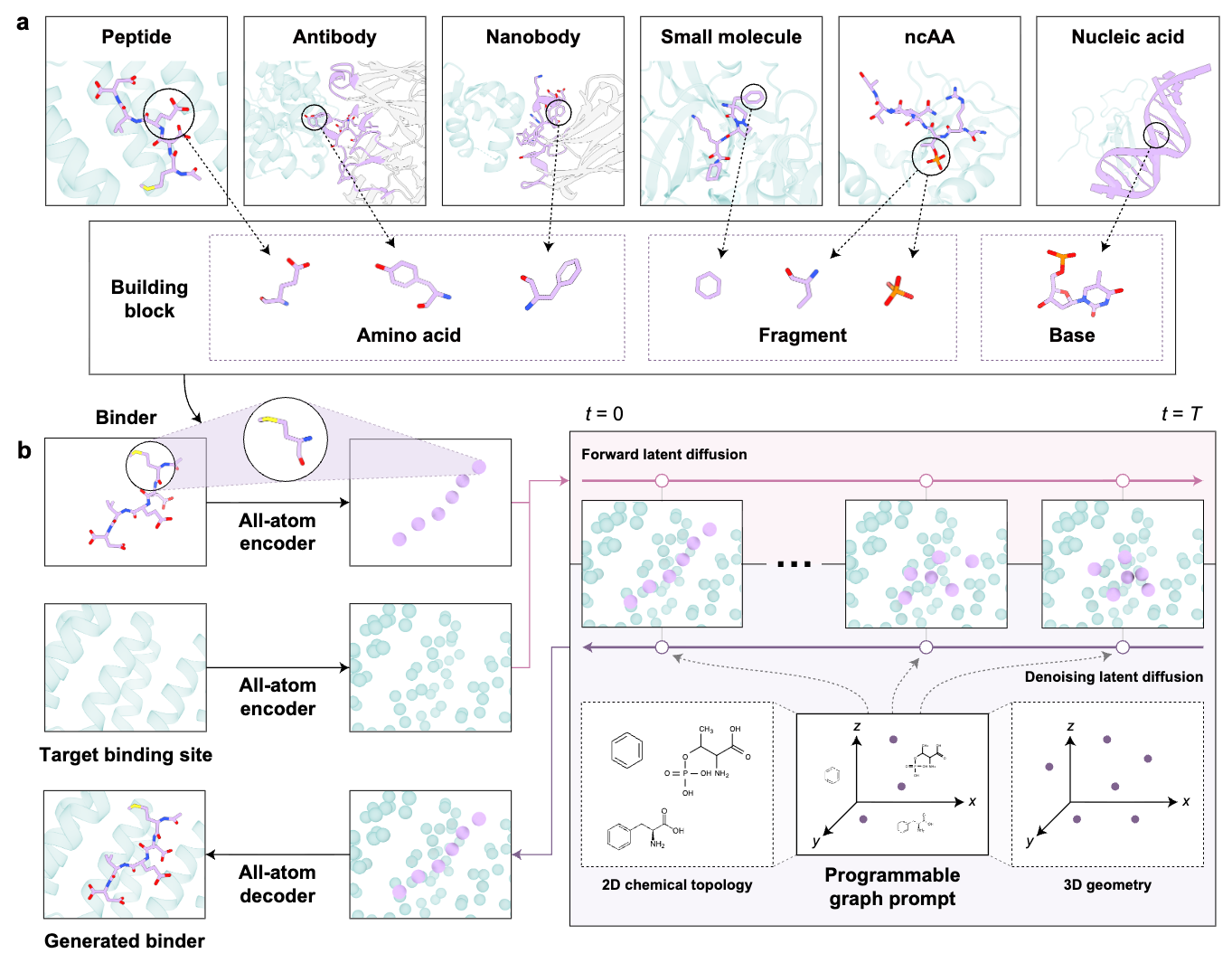
Biomolecular interactions lie at the core of cellular life, spanning diverse molecular modalities from small molecules to nucleic acids and proteins. Nevertheless, design strategies remain separated despite shared physicochemical principles of molecular recognition. Here we present AnewOmni, a unified generative framework trained on more than 5 million biomolecular complexes, that enables transferable molecular design across molecular scales by assembling chemically meaningful building blocks at atomic resolution. We further introduce programmable graph prompts to support user-defined chemical, topological, and geometric steering during generation, exploring hybrid and unconventional chemistries beyond canonical structures. We demonstrate that transferable learning of interaction patterns and physical constraints across molecular modalities is possible, via an atom-to-block latent space capturing both atomic details and structural priors. The framework successfully designed small molecules, peptides, and nanobodies targeting the challenging KRAS G12D switch II pocket, as well as orthosteric peptides and allosteric small-molecule inhibitors for PCSK9 in the absence of known binding site, achieving 23%-75% success with only low-throughput validation, bypassing modality-specific high-throughput screening. AnewOmni is the first to succeed in functional molecular design across all scales, from small chemical entities to large biologics, and represents a stepstone towards general molecular reasoning engines, advocating a generative foundation model for biomolecular interactions to enter regimes where data and human intuition remain limited.
Programming Biomolecular Interactions with All-Atom Generative Model
Xiangzhe Kong*, Junwei Chen*, Ziting Zhang*, Gaodeng Li*, Qingyuan Zhu, Lei Wei, Mingyu Li, Yan Shi, Weiyang Dai, Zishen Zhang, Wenjuan Tan, Rui Jiao, Xiaolun Wang, Jiqing Zheng, Ziyang Yu, Qilong Wu, Zhiye Guo, Li Zhang, Wentao Li, Qiaojing Huang, Xiaowo Wang, Wenbing Huang, Yuli She, Jian Zhang, Yang Liu, Kai Liu, Jianzhu Ma (* equal contribution)
Under review. 2026
Biomolecular interactions lie at the core of cellular life, spanning diverse molecular modalities from small molecules to nucleic acids and proteins. Nevertheless, design strategies remain separated despite shared physicochemical principles of molecular recognition. Here we present AnewOmni, a unified generative framework trained on more than 5 million biomolecular complexes, that enables transferable molecular design across molecular scales by assembling chemically meaningful building blocks at atomic resolution. We further introduce programmable graph prompts to support user-defined chemical, topological, and geometric steering during generation, exploring hybrid and unconventional chemistries beyond canonical structures. We demonstrate that transferable learning of interaction patterns and physical constraints across molecular modalities is possible, via an atom-to-block latent space capturing both atomic details and structural priors. The framework successfully designed small molecules, peptides, and nanobodies targeting the challenging KRAS G12D switch II pocket, as well as orthosteric peptides and allosteric small-molecule inhibitors for PCSK9 in the absence of known binding site, achieving 23%-75% success with only low-throughput validation, bypassing modality-specific high-throughput screening. AnewOmni is the first to succeed in functional molecular design across all scales, from small chemical entities to large biologics, and represents a stepstone towards general molecular reasoning engines, advocating a generative foundation model for biomolecular interactions to enter regimes where data and human intuition remain limited.
2025
GGFlow: A Graph Flow Matching Method with Efficient Optimal Transport
Xiaoyang Hou*, Milong Ren, Dongbo Bu, Xin Gao, Chunming Zhang, Shiwei Sun (* equal contribution)
AIDrugX Workshop, Neural Information Processing Systems (NeurIPS) 2024
Transactions on Machine Learning Research (TMLR) 2025
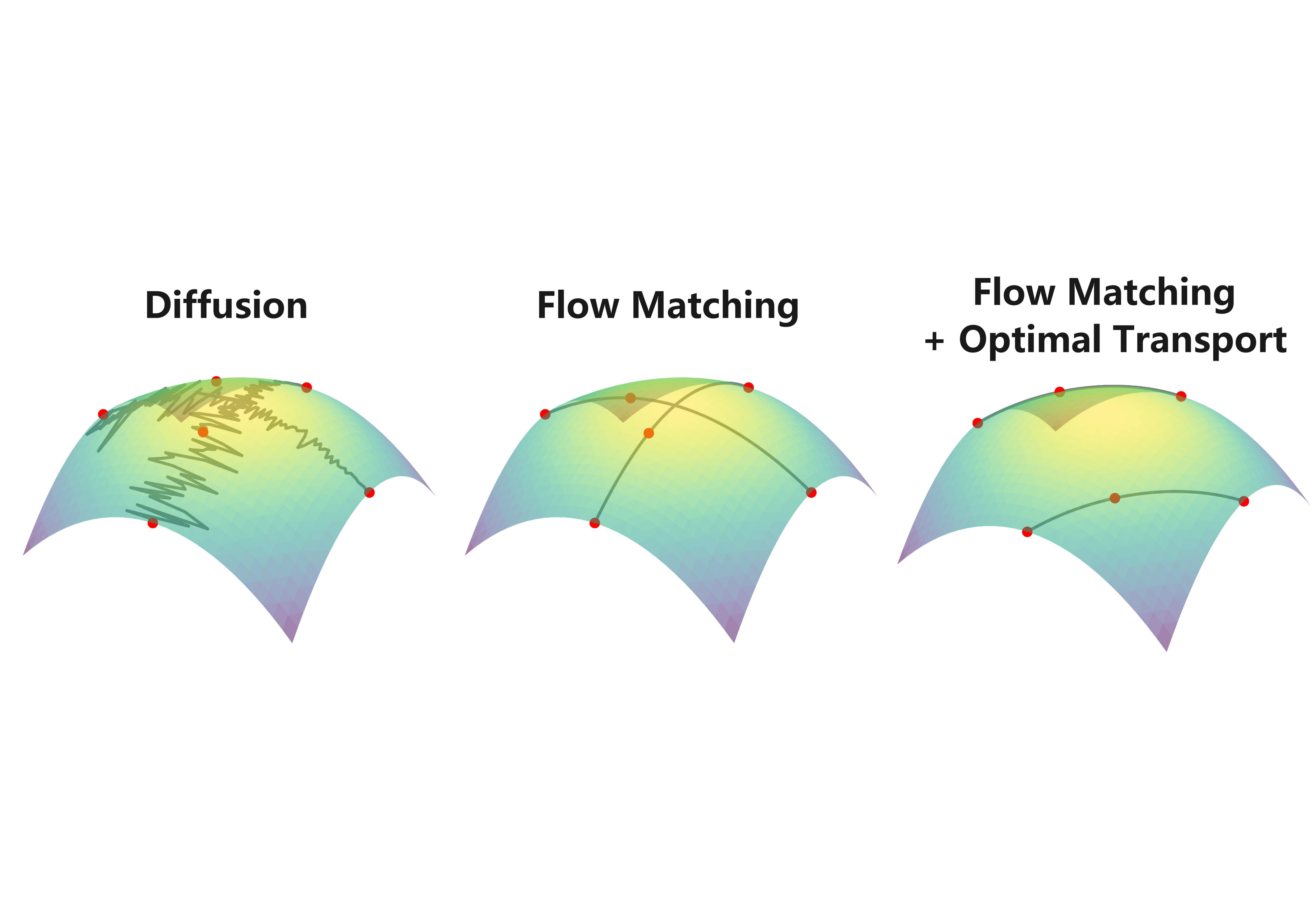
Generating graph-structured data is crucial in various domains but remains challenging due to the complex interdependencies between nodes and edges. While diffusion models have demonstrated their superior generative capabilities, they often suffer from unstable training and inefficient sampling. To enhance generation performance and training stability, we propose GGFlow, a discrete flow matching generative model incorporating an efficient optimal transport for graph structures and it incorporates an edge-augmented graph transformer to enable direct communications among edges. Additionally, GGFlow introduces a novel goal-guided generation framework to control the generative trajectory of our model towards desired properties. GGFlow demonstrates superior performance on both unconditional and conditional generation tasks, outperforming existing baselines and underscoring its effectiveness and potential for wider application.
GGFlow: A Graph Flow Matching Method with Efficient Optimal Transport
Xiaoyang Hou*, Milong Ren, Dongbo Bu, Xin Gao, Chunming Zhang, Shiwei Sun (* equal contribution)
AIDrugX Workshop, Neural Information Processing Systems (NeurIPS) 2024
Transactions on Machine Learning Research (TMLR) 2025
Generating graph-structured data is crucial in various domains but remains challenging due to the complex interdependencies between nodes and edges. While diffusion models have demonstrated their superior generative capabilities, they often suffer from unstable training and inefficient sampling. To enhance generation performance and training stability, we propose GGFlow, a discrete flow matching generative model incorporating an efficient optimal transport for graph structures and it incorporates an edge-augmented graph transformer to enable direct communications among edges. Additionally, GGFlow introduces a novel goal-guided generation framework to control the generative trajectory of our model towards desired properties. GGFlow demonstrates superior performance on both unconditional and conditional generation tasks, outperforming existing baselines and underscoring its effectiveness and potential for wider application.
2024
Accurate structure prediction of immune proteins using parameter-efficient transfer learning
Milong Ren*, Zaikai He, Siyuan Tao, Ming Li, Dongbo Bu, Haicang Zhang (* equal contribution)
BioRxiv 2024
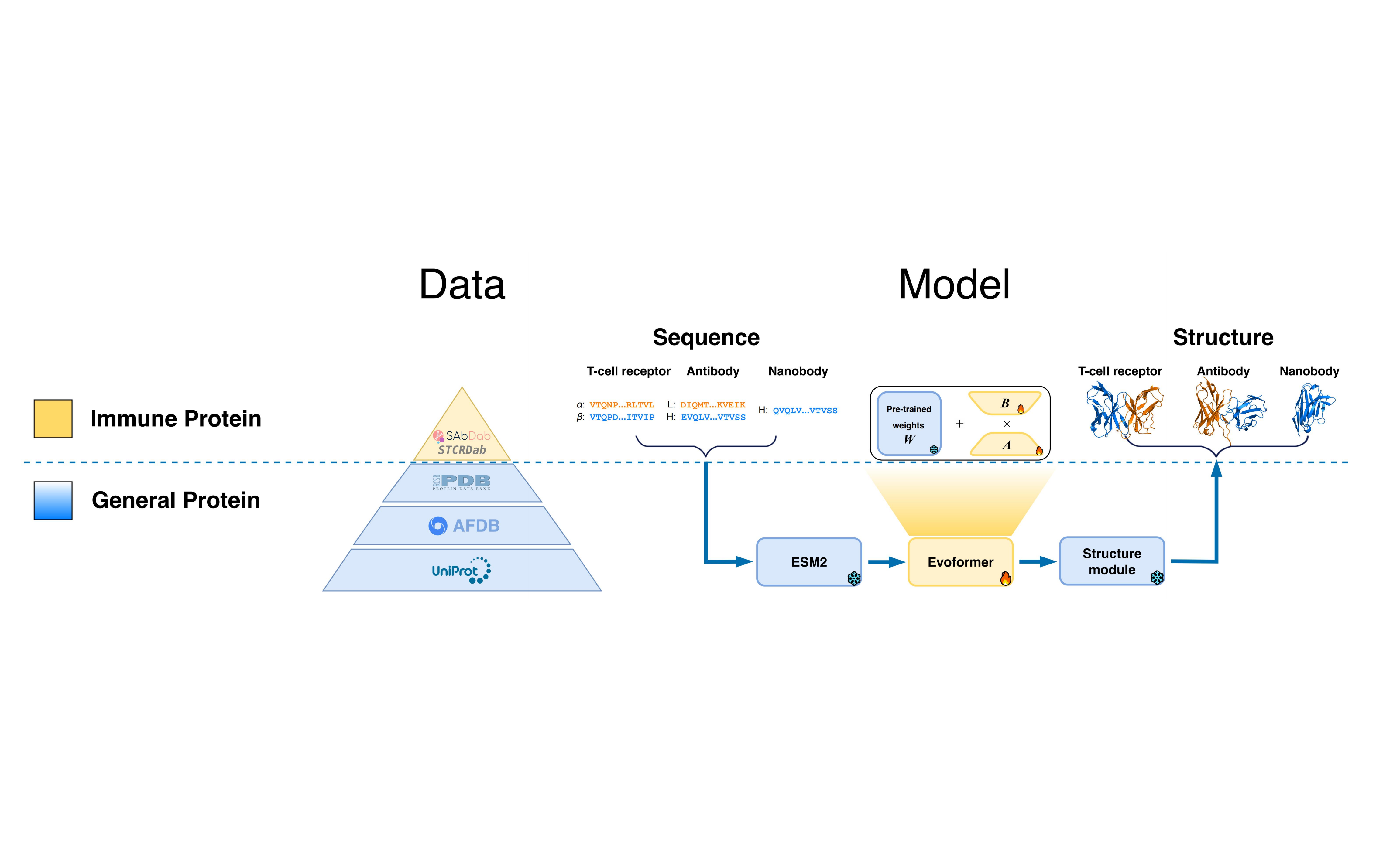
We propose ImmuneFold, a transfer learning approach that fine-tunes ESMFold specifically for immune proteins. We leverage low-rank adaption (LoRA), a parameter-efficient fine-tuning technique that requires considerably less memory and substantially fewer parameters. Evaluations on various immune proteins, including T-cell receptors, antibodies, and nanobodies, demonstrate that ImmuneFold outperforms existing methods in prediction accuracy. Furthermore, we apply ImmuneFold to develop a zero-shot protocol for TCR-epitope binding prediction. Unlike previous supervised methods suffering from severe overfitting due to limited experimental binding data, our approach first predicts TCR-epitope structure using ImmuneFold and then directly estimates the binding affinity by calculating Rosseta energy.
Accurate structure prediction of immune proteins using parameter-efficient transfer learning
Milong Ren*, Zaikai He, Siyuan Tao, Ming Li, Dongbo Bu, Haicang Zhang (* equal contribution)
BioRxiv 2024
We propose ImmuneFold, a transfer learning approach that fine-tunes ESMFold specifically for immune proteins. We leverage low-rank adaption (LoRA), a parameter-efficient fine-tuning technique that requires considerably less memory and substantially fewer parameters. Evaluations on various immune proteins, including T-cell receptors, antibodies, and nanobodies, demonstrate that ImmuneFold outperforms existing methods in prediction accuracy. Furthermore, we apply ImmuneFold to develop a zero-shot protocol for TCR-epitope binding prediction. Unlike previous supervised methods suffering from severe overfitting due to limited experimental binding data, our approach first predicts TCR-epitope structure using ImmuneFold and then directly estimates the binding affinity by calculating Rosseta energy.
GTAM: A Molecular Pretraining Model with Geometric Triangle Awareness
Xiaoyang Hou*, Milong Ren*, Bo Duan, Chunming Zhang, Dongbo Bu, Shiwei Sun (* equal contribution)
Bioinformatics 2024
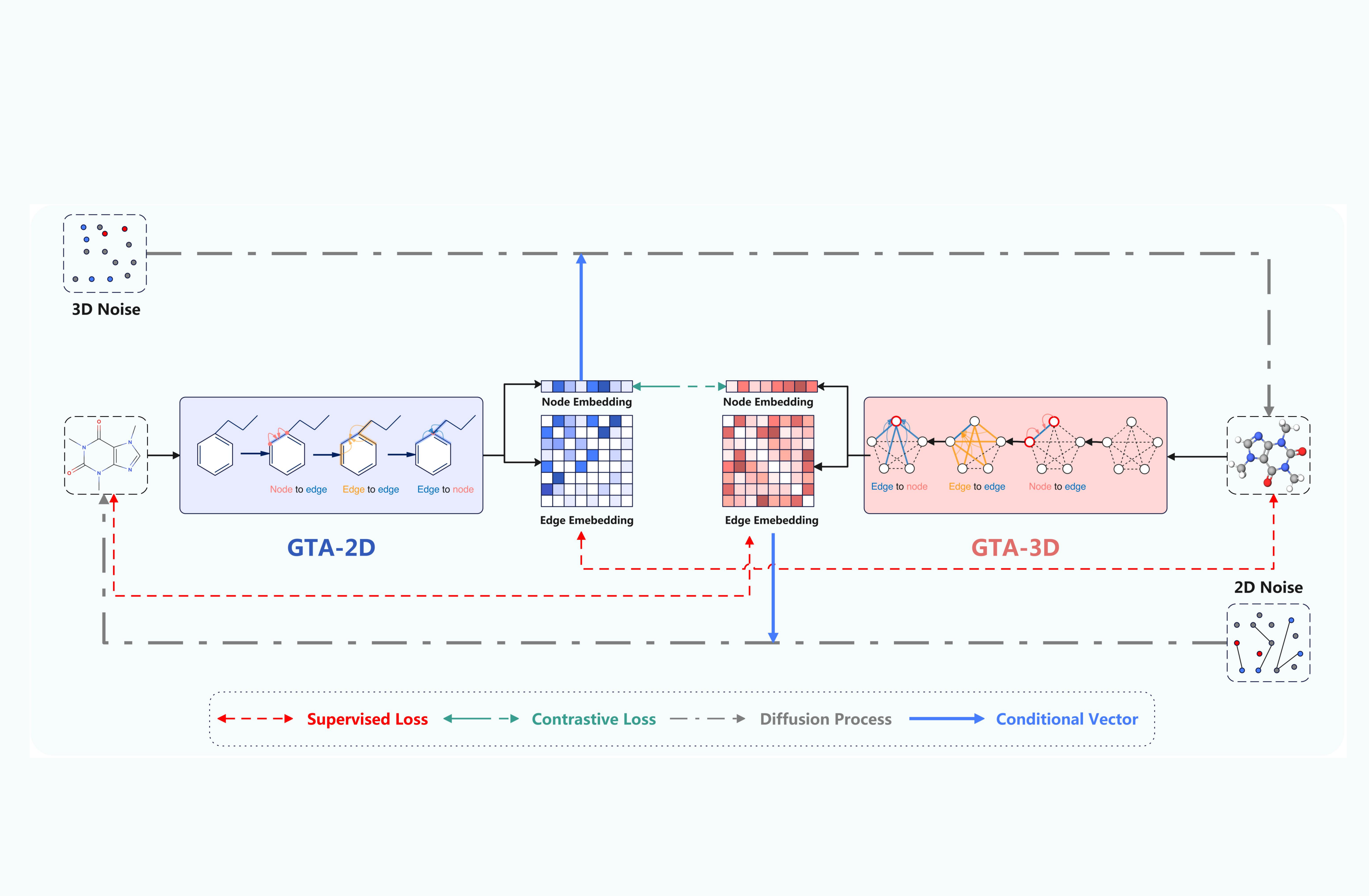
This method integrates innovative molecular encoders for both 2D graphs and 3D conformations, enabling the accurate capture of geometric dependencies among edges in graph-based molecular structures. Furthermore, GTAM is bolstered by the development of two contrastive training objectives designed to facilitate the direct transfer of edge information between 2D topological graphs and 3D geometric conformations, enhancing the functionality of the molecular encoders.
GTAM: A Molecular Pretraining Model with Geometric Triangle Awareness
Xiaoyang Hou*, Milong Ren*, Bo Duan, Chunming Zhang, Dongbo Bu, Shiwei Sun (* equal contribution)
Bioinformatics 2024
This method integrates innovative molecular encoders for both 2D graphs and 3D conformations, enabling the accurate capture of geometric dependencies among edges in graph-based molecular structures. Furthermore, GTAM is bolstered by the development of two contrastive training objectives designed to facilitate the direct transfer of edge information between 2D topological graphs and 3D geometric conformations, enhancing the functionality of the molecular encoders.
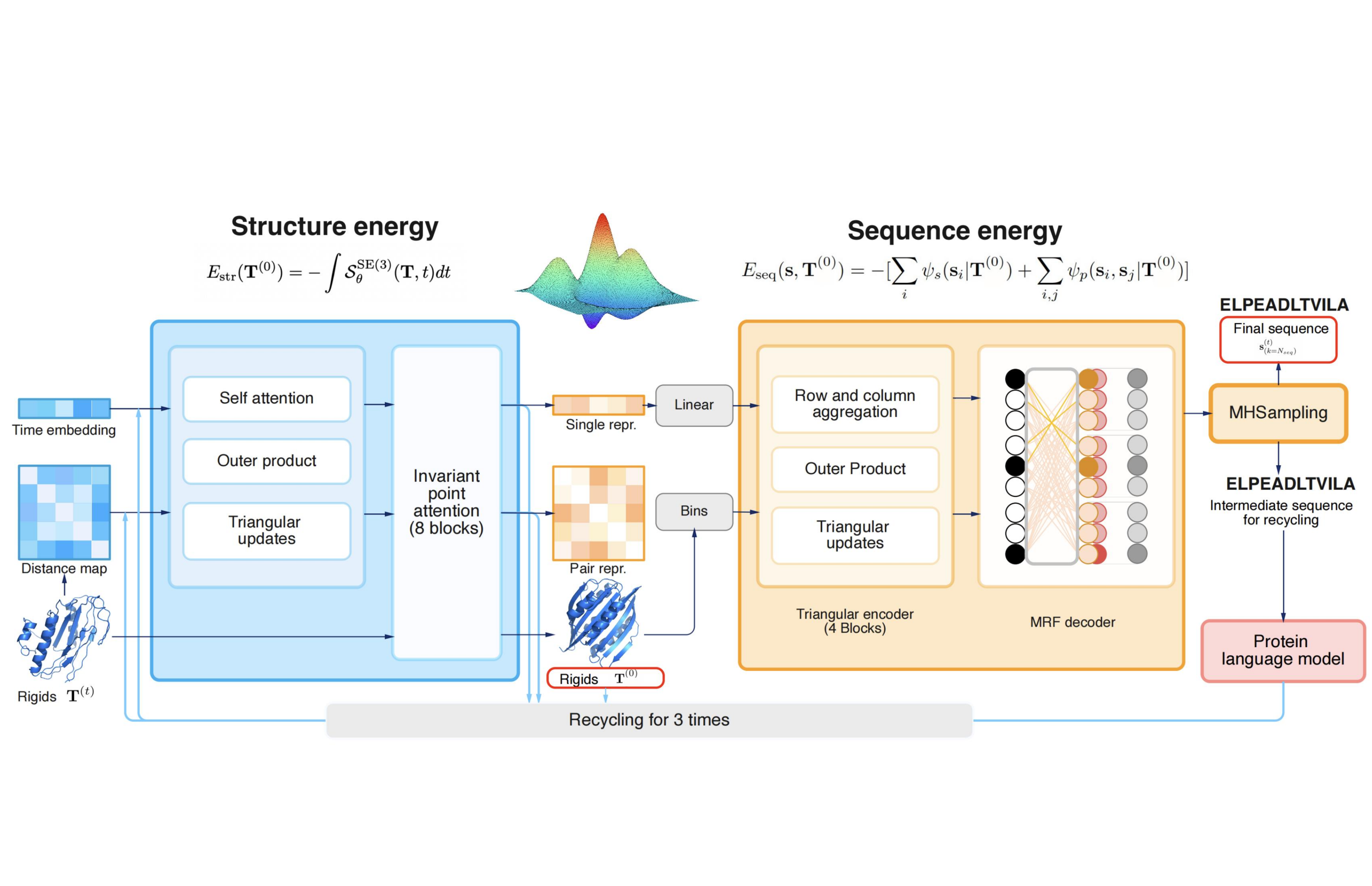
CarbonNovo: Joint Design of Protein Structure and Sequence Using a Unified Energy-based Model
Milong Ren, Haicang Zhang
International Conference on Machine Learning (ICML) 2024
We propose CarbonNovo, a unified energy-based model for jointly generating protein structure and sequence. Specifically, we leverage a score-based generative model and Markov Random Fields for describing the energy landscape of protein structure and sequence. In CarbonNovo, the structure and sequence design module communicates at each diffusion step, encouraging the generation of more coherent structure-sequence pairs. Moreover, the unified framework allows for incorporating the protein language models as evolutionary constraints for generated proteins.
CarbonNovo: Joint Design of Protein Structure and Sequence Using a Unified Energy-based Model
Milong Ren, Haicang Zhang
International Conference on Machine Learning (ICML) 2024
We propose CarbonNovo, a unified energy-based model for jointly generating protein structure and sequence. Specifically, we leverage a score-based generative model and Markov Random Fields for describing the energy landscape of protein structure and sequence. In CarbonNovo, the structure and sequence design module communicates at each diffusion step, encouraging the generation of more coherent structure-sequence pairs. Moreover, the unified framework allows for incorporating the protein language models as evolutionary constraints for generated proteins.
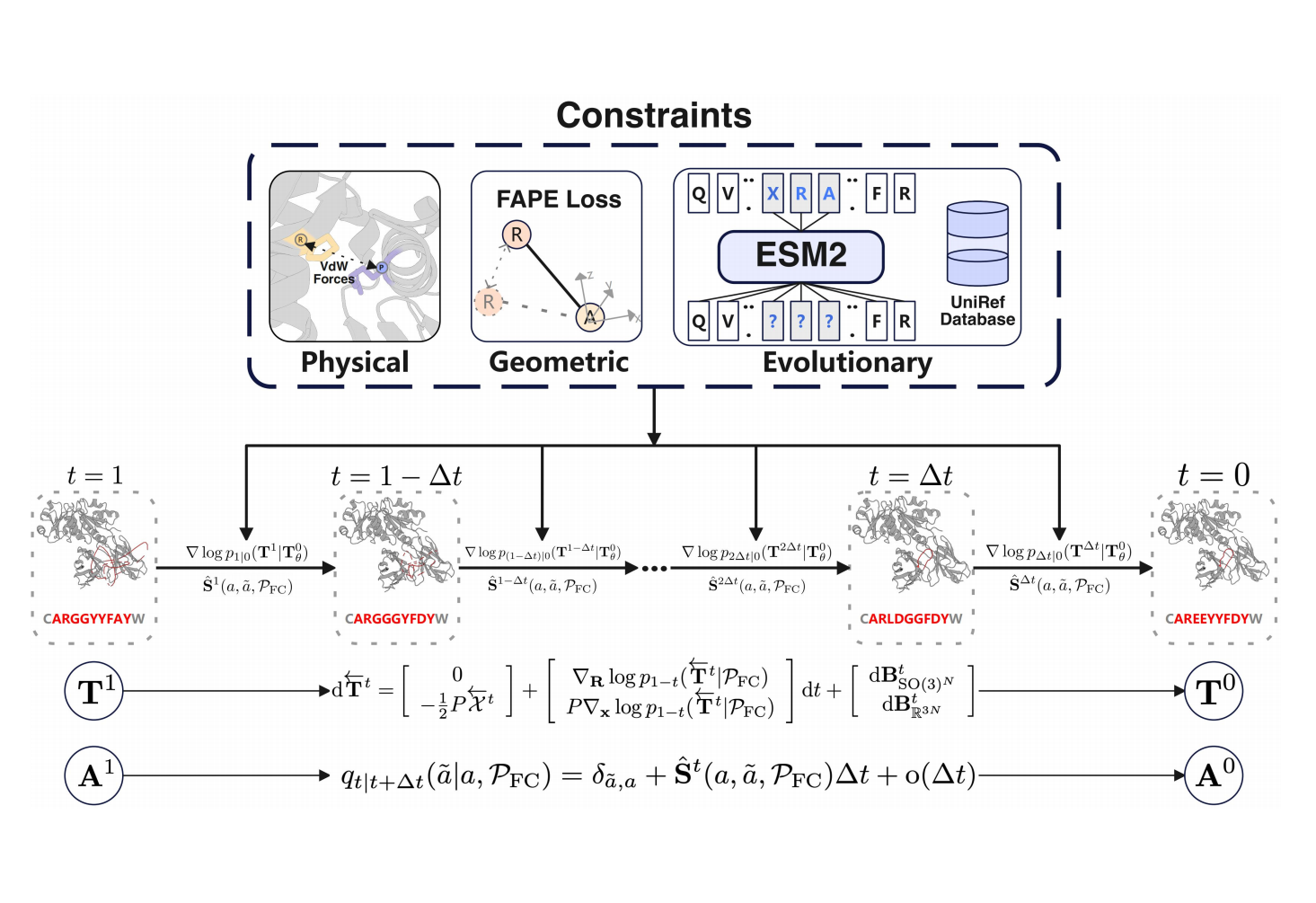
Antibody Design Using a Score-based Diffusion Model Guided by Evolutionary, Physical and Geometric Constraints
Milong Ren, Haicang Zhang
International Conference on Machine Learning (ICML) 2024
We present AbX, a new score-based diffusion generative model guided by evolutionary, physical, and geometric constraints for antibody design. These constraints serve to narrow the search space and provide priors for plausible antibody sequences and structures. Specifically, we leverage a pre-trained protein language model as priors for evolutionary plausible antibodies and introduce additional training objectives for geometric and physical constraints like van der Waals forces. Furthermore, as far as we know, AbX is the first score-based diffusion model with continuous timesteps for antibody design, jointly modeling the discrete sequence space and the $SE(3)$ structure space.
Antibody Design Using a Score-based Diffusion Model Guided by Evolutionary, Physical and Geometric Constraints
Milong Ren, Haicang Zhang
International Conference on Machine Learning (ICML) 2024
We present AbX, a new score-based diffusion generative model guided by evolutionary, physical, and geometric constraints for antibody design. These constraints serve to narrow the search space and provide priors for plausible antibody sequences and structures. Specifically, we leverage a pre-trained protein language model as priors for evolutionary plausible antibodies and introduce additional training objectives for geometric and physical constraints like van der Waals forces. Furthermore, as far as we know, AbX is the first score-based diffusion model with continuous timesteps for antibody design, jointly modeling the discrete sequence space and the $SE(3)$ structure space.
2023
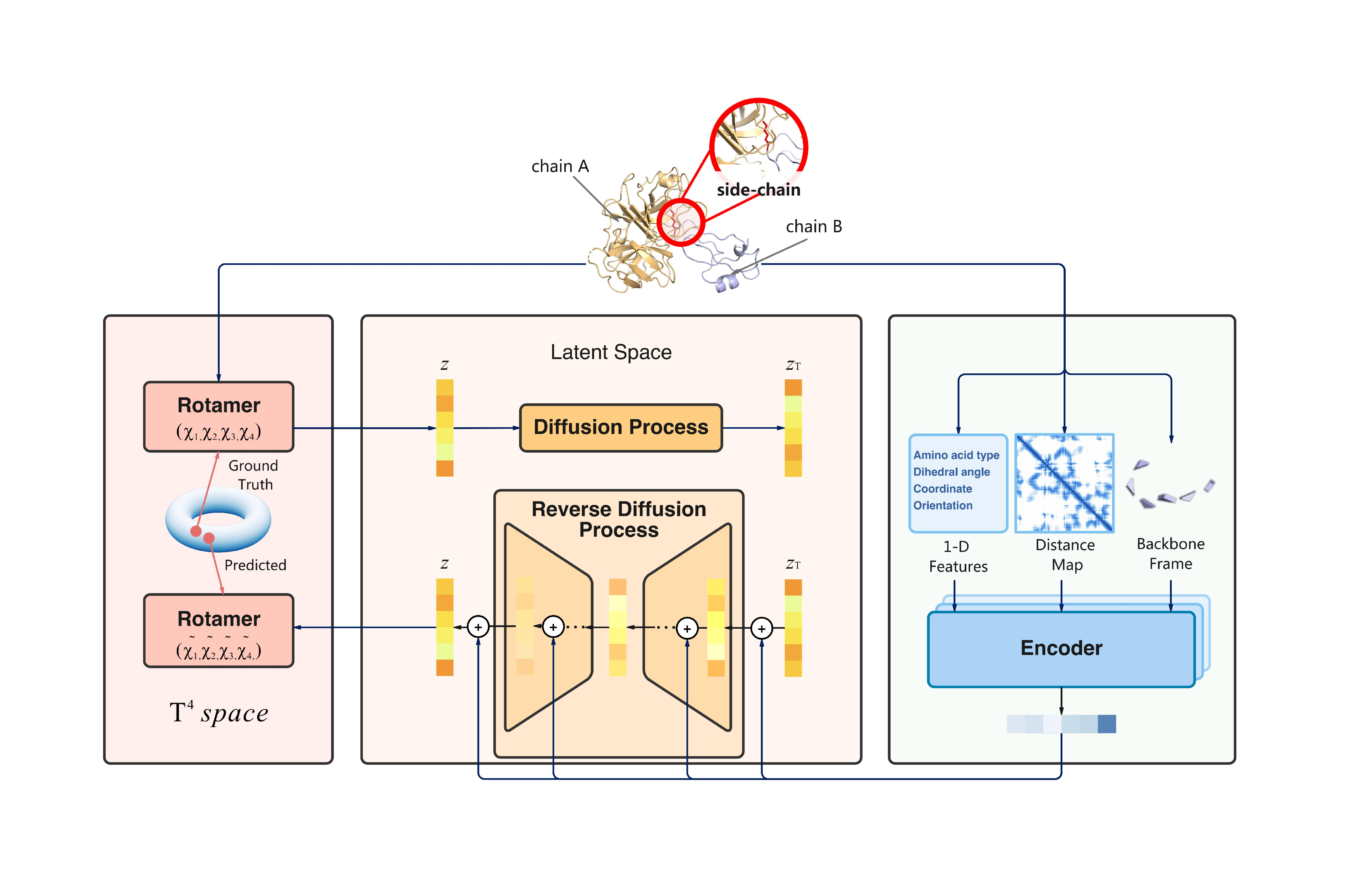
Predicting mutational effects on protein-protein binding via a side-chain diffusion probabilistic model
Shiwei Liu*, Milong Ren, Chungong Yu, Dongbo Bu, Haicang Zhang (* equal contribution)
Neural Information Processing Systems (NeurIPS) 2023
In this work, we propose SidechainDiff, a representation learning-based approach that leverages unlabelled experimental protein structures. SidechainDiff utilizes a Riemannian diffusion model to learn the generative process of side-chain conformations and can also give the structural context representations of mutations on the protein-protein interface.
Predicting mutational effects on protein-protein binding via a side-chain diffusion probabilistic model
Shiwei Liu*, Milong Ren, Chungong Yu, Dongbo Bu, Haicang Zhang (* equal contribution)
Neural Information Processing Systems (NeurIPS) 2023
In this work, we propose SidechainDiff, a representation learning-based approach that leverages unlabelled experimental protein structures. SidechainDiff utilizes a Riemannian diffusion model to learn the generative process of side-chain conformations and can also give the structural context representations of mutations on the protein-protein interface.